
28
Apr
Rare examples of mutations at the prosaposin or activator locus also lead to severe Gaucher disease-like phenotypes with glucosylceramide accumulation. Imiglucerase significantly improves BMD in patients with GD with 8.
Effects of gaucher disease. The results indicated that bone pain and chronic fatigue interfered with school job and social activities and were the most debilitating symptoms of Gaucher disease. Most patients experienced a significant increase in energy level from therapy and reported significant improvements in quality of life. Most patients did not perceive an effect of Gaucher disease on their overall emotional health but some.
What are the complications of Gaucher disease. Gaucher disease can cause other health problems such as. Trouble walking or getting around.
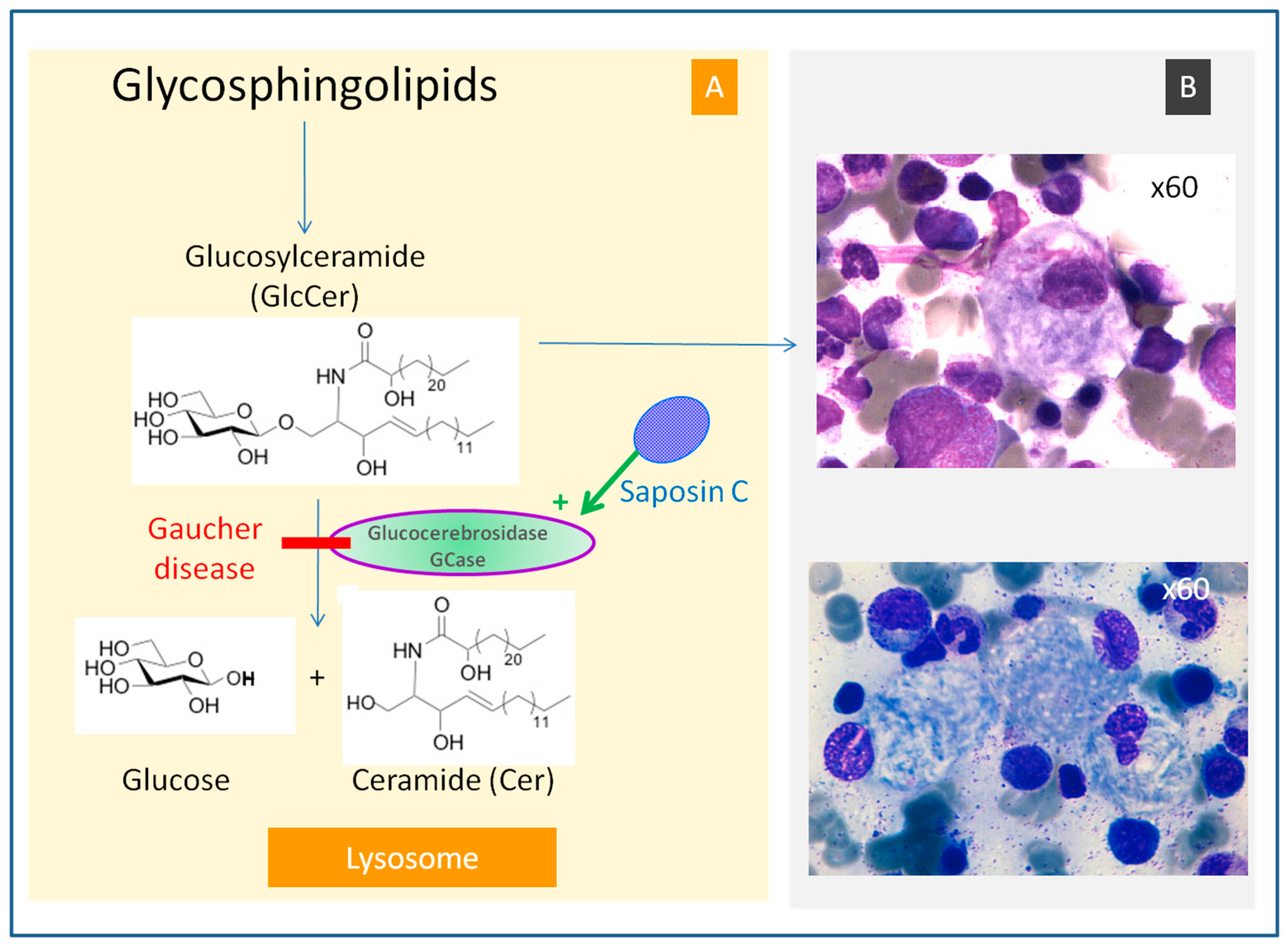
Not having enough healthy red blood cells anemia Extreme tiredness fatigue What can I do to prevent Gaucher disease. Gaucher disease is a lysosomal storage disease resulting from glycocerebroside accumulation in macrophages due to a genetic deficiency of the enzyme glucocerebrosidase. It may occur in adults but occurs most severely in infants in whom cerebroside also accumulates in neurons.
Patients with Gauchers disease experience enlargement of the liver and spleen and bone destruction. Gaucher disease can cause severe arthritis and joint damage which can be permanent if the disease is untreated. Gaucher disease treatment is key to avoiding permanent damage to your body.
Testing for Gaucher disease involves a blood test called a beta-glucosidase leukocyte BGL test to determine enzyme levels in your body. Its impact in rare disease populations such as Gaucher disease GD is unknown. In GD decreased acid β-glucosidase activity leads to the accumulation of inflammatory glycosphingolipids and chronic myeloid cell immune activation which a priori could predispose to the most severe effects of SARS-CoV-2.
To evaluate the determinants of SARS-CoV-2 infection in GD we conducted a cross-sectional. The effects of phosphatidylserine on glucocerebrosidase from normal and Gaucher fibroblasts. Glucocerebroside beta-glucosidase glucocerebrosidase activity was assayed from cultured fibroblasts of normal individuals and patients with type 1 non-neuropathic type 2 acute neuropathic and type 3 subacute neuropathic form of Gaucher disease.
Gaucher disease GD arises because of defective acid β-glucosidase glucosylceramide accumulates in mononuclear phagocytes causing hepatosplenomegaly. The three types of GD are non-neuronopathictype I acute neuronopathic or GDtype II and subacute neuronopathic or GDtype III. Gaucher Disease Type 1 Symptoms Patients with Gaucher disease dont have enough glucocerebrosidase GCase an enzyme that breaks down a fatty chemical in the body called glucocerebroside.
When glucocerebroside builds up in the organs and bone marrow it causes signs and symptoms such as. Gaucher disease is the commonest lysosomal storage disease seen in India and worldwide. It should be considered in any child or adult with an unexplained splenohepatomegaly and cytopenia which are seen in the three types of Gaucher disease.
Type 1 is the non-neuronopathic form and type 2 and 3 are the neuronopathic forms. The main signs and symptoms of Gaucher disease include the following. Anemia low red blood cell count Fatigue tiredness Low platelet count that can lead to easy bruising Enlarged spleen and liver hepatosplenomegaly Easy bleeding that is difficult to stop Bone pain bone crisis severe bone.
Symptoms may begin in early life or adulthood and include skeletal disorders and bone lesions that may cause pain and fractures enlarged spleen and liver liver malfunction anemia and yellow spots in the eyes. There are three common clinical subtypes of Gaucher disease. Gaucher disease is associated with an elevated risk of developing malignancies such as liver cancer and various forms of blood cancer.
The adverse effects of this disease on the skeletal system can produce severe arthritis and even weaken the bones to the extent of. The variant GCases from Gaucher disease patients and selected variant GCases from the mouse had decreased relative k cat and differential effects on active site binding andor attachment of mechanism-based covalent conduritol B epoxide or reversible deoxynojirimycin derivatives inhibitors. A defect in negatively charged phospholipid activation was present in the majority of variant GCases.
Imiglucerase significantly improves BMD in patients with GD with 8. Years of ERT leading to normal BMD. IntroductionThe objective was to determine the effect of enzyme replacement therapy ERT.
Cerezyme imiglucerase on BMD in type 1 Gaucher disease GD. Deficiency of the lysosomal enzyme glucocerebrosidase leads to the accumulation of glucocerebroside in spleen liver and bone marrow. 12 In the early 1990s Gaucher disease was the first of the lysosomal storage disorders that could be treated successfully with enzyme replacement therapy ERT using mannose-terminated enzyme from placental tissue alglucerase or recombinant.
These autosomal recessively inherited disorders result almost exclusively from mutations in the gene encoding the lysosomal hydrolase acid β-glucosidase or glucocerebrosidase EC 32145. Rare examples of mutations at the prosaposin or activator locus also lead to severe Gaucher disease-like phenotypes with glucosylceramide accumulation.
Previous post
Effects of hypothyroidism in malesNext post
Effects of fiber in the diet